The calculation of the electron–phonon coupling from frst principles is computationally very challenging and remains mostly out of reach for systems with a large number of atoms. Semi-empirical methods, like density functional tight binding (DFTB), provide a framework for obtaining quantitative results at moderate computational costs. Herein, we present a new method based on the DFTB approach for computing electron–phonon couplings and relaxation times. It interfaces with phonopy for vibrational modes and dftb+ to calculate transport properties. We derive the electron–phonon coupling within a non-orthogonal tight-binding framework and apply them to graphene as a test case.
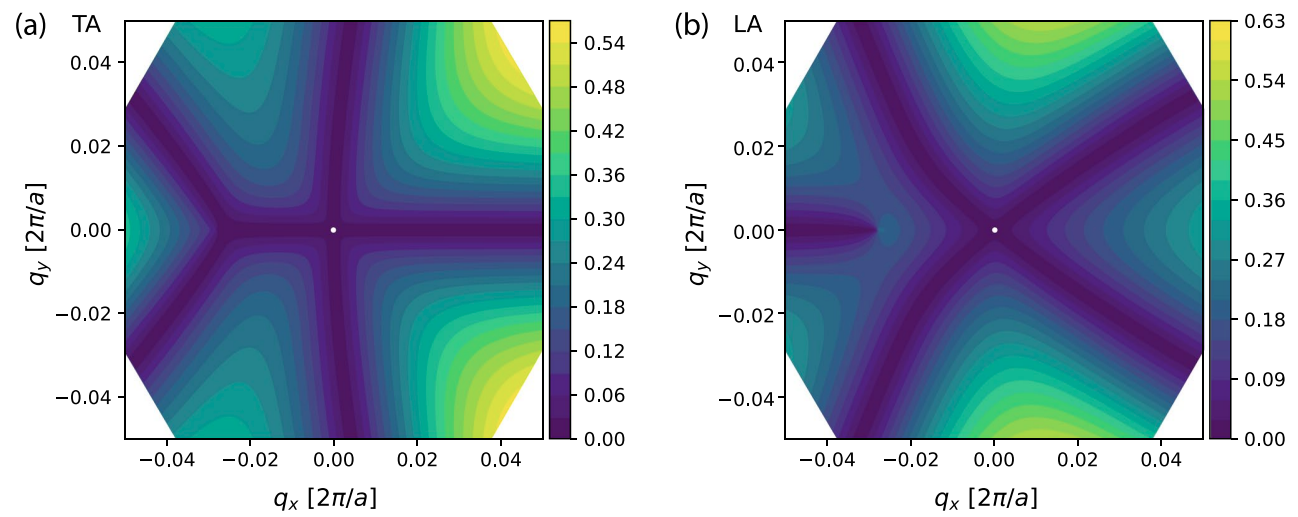
The calculation of the electron–phonon coupling from frst principles is computationally very challenging and remains mostly out of reach for systems with a large number of atoms. Semi-empirical methods, like density functional tight binding (DFTB), provide a framework for obtaining quantitative results at moderate computational costs. Herein, we present a new method based on the DFTB approach for computing electron–phonon couplings and relaxation times. It interfaces with phonopy for vibrational modes and dftb+ to calculate transport properties. We derive the electron–phonon coupling within a non-orthogonal tight-binding framework and apply them to graphene as a test case.



